Uncertainty quantification of a newly optimized methanol/NOx combustion mechanism Márton Kovács, Máté Papp, István Gy. Zsély, Tibor Nagy,
Tamás Turányi
Proc.Combust.Inst., 41,
105938 (2025)

A comprehensive uncertainty analysis was conducted on a recently proposed
optimized detailed methanol/NOx combustion mechanism, focusing on its predictions for the experimental data used as targets in the optimization.
The primary sources of model uncertainty in both the initial and the optimized mechanisms were compared. The propagation of uncertainties in
kinetic and thermodynamic model parameters to the simulation results was investigated using approaches of varying complexity. Both absolute model
uncertainties and, as a novelty, those normalized by experimental uncertainties were considered. The effect of correlation among the Arrhenius
parameters in optimized reactions was examined through local uncertainty analysis. Accounting for parameter correlations yielded a more accurate
representation of model uncertainty, although using the temperature-average of the uncertainty parameters also provided a reasonable approximation. The
impact of correlations among all kinetic parameters was assessed using global uncertainty analysis with Monte Carlo sampling, which supported these
conclusions. The analyses demonstrated that parameter optimization can significantly reduce model uncertainty. On average, the root-mean-square
model uncertainty, normalized by the experimental uncertainty, decreased from a factor of 5.5 to 2.4 upon optimization. The dominant uncertainty
contributions from the CH
2O + NO
2 = HONO +HCO and CH
3OH
+ NO
2 = HONO + CH
2OH reactions were effectively
eliminated in the process. However, reactions involving the CH
2OH
radical with NO
2, NO, HNO, and O
2 remained significant
sources of uncertainty. To further reduce the model uncertainty, future research should focus on these reactions. This includes indirect
experimental measurements sensitive to these pathways, as well as direct measurements or theoretical calculations of their rate coefficients.
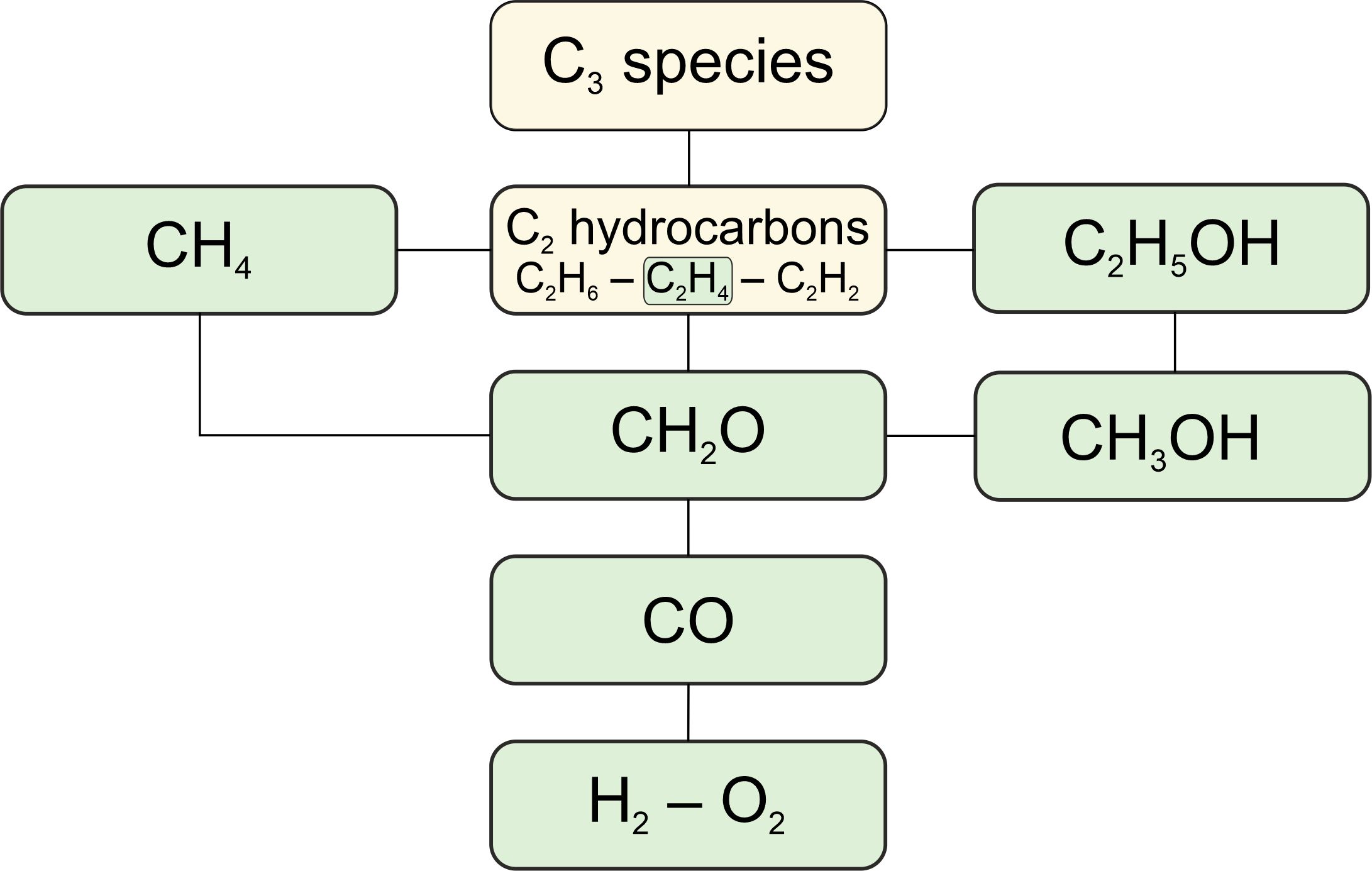
Comparison of methane combustion mechanisms using concentration measurements Peng Zhang, István Gyula Zsély, Máté Papp, Ákos Veres-Ravai, Boyang Su, Tibor Nagy, Bin Yang, Tamás Turányi
Combustion and Flame, 282,
114499 (2025)

A
large amount of concentration measurement data from combustion
experiments carried out in flow reactors, jet-stirred reactors, and
shock tubes for methane (+ H2/CO)-oxygen-diluent mixtures covering wide
ranges of temperature, pressure, equivalence ratio, and diluent ratio
were collected from 45 publications. Altogether, 35420 concentration
data points in 965 datasets were encoded in 332 XML data files and made
available on the ReSpecTh site (https://respecth.hu). The measured
species are CH4, C2H2, C2H4, C2H6, aC3H4 (allene), pC3H4 (propyne), C3H6
(propene), C3H8, C4H2 (diacetylene), C4H4 (vinylacetylene), C6H6
(benzene), CH2O, CH3OH, C2H5OH, O2, CO, CO2, H2, and H2O. In some shock
tube measurements, the concentrations of radicals CH, CH3, and OH were
also measured. The diluents were N2, H2O, CO2, Ar and He. The
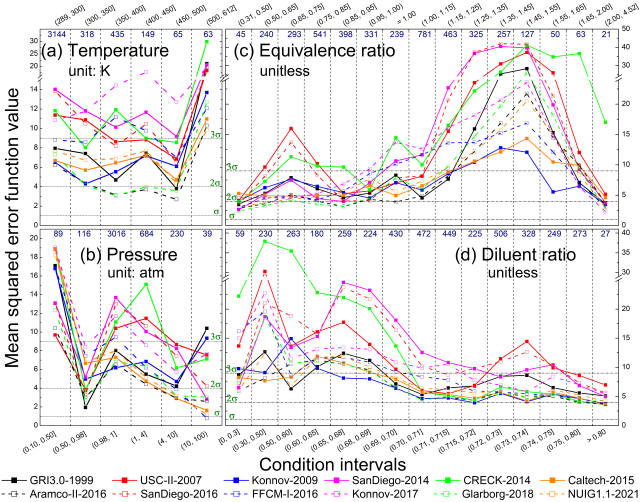
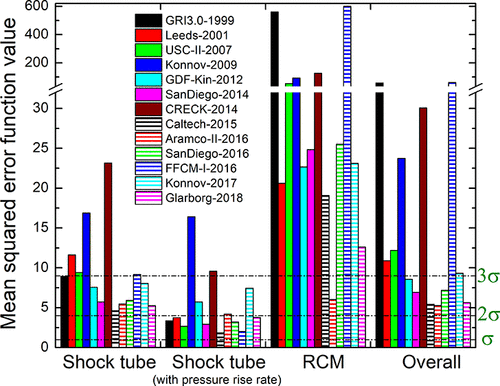
performance of 13 methane combustion mechanisms was analysed based on
their capability to reproduce concentration measurements across various
experimental types and conditions. A novel consideration of temperature
uncertainty was proposed and applied in the performance evaluation of
models. The mechanisms Aramco-II-2016, Konnov-2017, NUIG1.1-2021,
Glarborg-2018, Caltech-2015, and FFCM-I-2016 were the best-performing
mechanisms in ascending order of error. Aramco-II-2016 reproduces the
concentrations of flow reactors the best, especially for CH4, O2, CO,
CO2, CH3OH, and C2 hydrocarbons. Konnov-2017 is good at predicting
concentrations in shock tubes, and FFCM-I-2016 has the overall lowest
errors for helium-containing measurements. Local sensitivity analysis
based on the Aramco-II-2016 mechanism identified 42 important elementary
reactions. Directly measured and theoretically calculated rate
coefficients (1879 data points in 133 datasets) were collected for these
elementary reactions and compared with those used in the investigated
methane combustion mechanisms.
Continue reading...
ReSpecTh: Reaction kinetics, spectroscopy, and thermochemical datasets
Tamás Turányi, István Gy. Zsély, Máté Papp, Tibor Nagy, Tibor Furtenbacher, Roland Tóbiás, Péter Árendás, Attila G. Császár
Scientific Data, 12,
1021 (2025)
Description
of a large number of datasets related to gas-phase reaction kinetics
(Re), high-resolution molecular spectroscopy (Spec), and thermochemistry
(Th), called ReSpecTh, is presented. The datasets contain accurate and
validated experimental, empirical, and computed, machine-searchable
data, and, whenever possible, the corresponding metadata. ReSpecTh data
and the accompanying utility codes can be used in several engineering
and scientific fields either separately or simultaneously, such as
simulation of combustion reactions, atmospheres of planets and
exoplanets, and stellar and interstellar environments.
Continue reading...
Determination of key rate parameters of the Thermal DeNOx process by optimization of a detailed combustion kinetic mechanism András György Szanthoffer, Máté Papp, Peter Glarborg, Hamid Hashemi, István Gyula Zsély, Tamás Turányi
Int. J. Chem. Kinet., 57, 434-445 (2025)
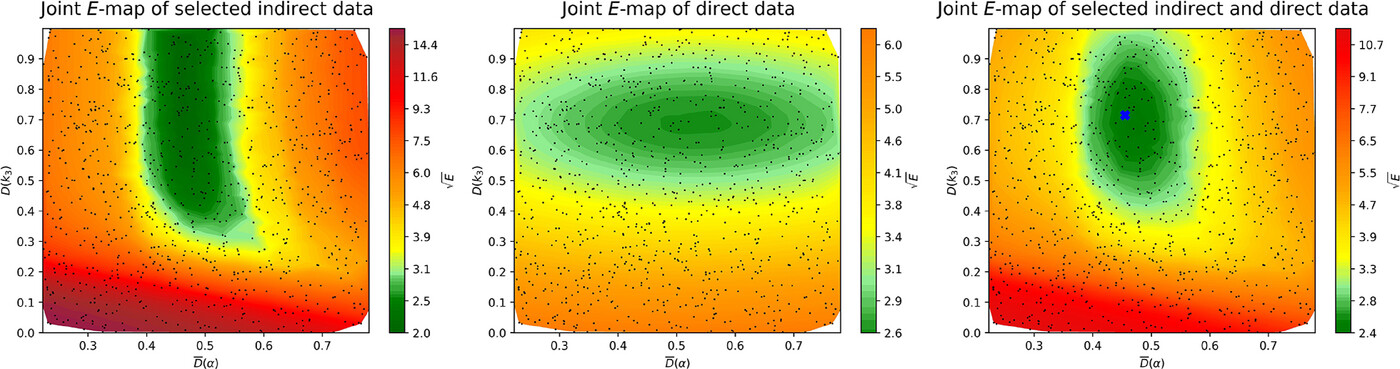
The thermal DeNOx process is a widely used NOx emission control technique,
but its chemical kinetic description still lacks accuracy. In the present work, two key kinetic parameters of the thermal DeNOx process were
investigated: the branching fraction (α) of the NH2 + NO
reaction, and the rate coefficient of the unimolecular decomposition of NNH (inverse lifetime of NNH, τNNH). Values of these
rate parameters were determined using a mechanism optimization method that utilizes both direct and indirect data and minimizes the value of an error
function. Data were collected from the literature and available in data files on the ReSpecTh site (https://ReSpecTh.hu). Indirect experimental
data used as optimization targets were NO mole fractions measured in tubular flow reactors. The most recent nitrogen chemistry mechanism of
Glarborg and coworkers (2024) was used as the initial mechanism. Inconsistency was found between the indirect experimental data, and
therefore mechanism optimization was not feasible using all indirect data. Using a new algorithm, a consistent subset of indirect data was
identified. The optimized value of τNNH
(8.5 ∙ 10−11 s) is approximately an order of
magnitude smaller than in the initial mechanism (10−9 s),
but consistent with theoretical calculations. The posterior uncertainty of τNNH is significantly smaller than its prior
uncertainty. The optimized value of the branching fraction is different from its initial value by less than 2%, but due to the very large
sensitivity of the simulation results to α, this small change
improves the performance of the mechanism noticeably. The width of the posterior uncertainty range of α is approximately half that of its
prior uncertainty range, estimated using only direct measurements. This is a significant improvement, but more accurate indirect experimental data
are needed to further increase the accuracy of the determination of α.
Continue reading...
Alnasif A, Jójka J, Papp M, Szanthoffer AG, Kovaleva M, Turányi T, Mashruka S, Valera-Medina A, Nagy T
The Journal of Ammonia Energy, 03, 054–072 (2025)
https://doi.org/10.18573/jae.46 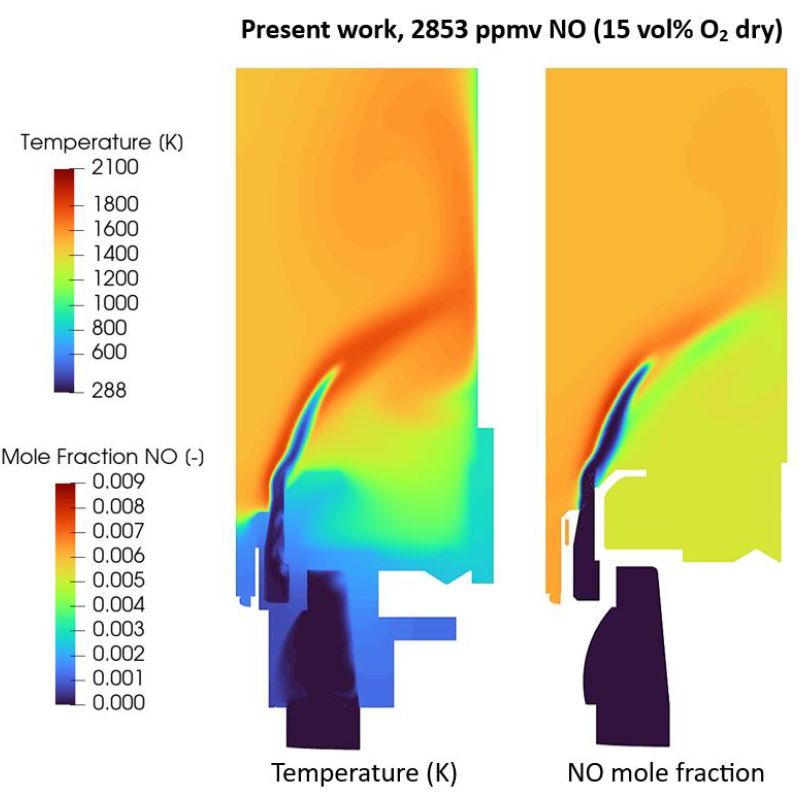
Ammonia (NH3) has been considered a potential
fuel for energy production to achieve zero carbon emissions. However,
several challenges must be addressed to ensure its widespread use and
safety. The current work focuses on developing a kinetic reaction
mechanism that not only accurately predicts laminar flame speeds and the
emissions from NH3 and NH3/H2 flames across various conditions but also
ensures seamless applicability in Computational Fluid Dynamics (CFD)
simulations, particularly in scenarios involving turbulent flows, such
as swirl burners or complex engine chamber conditions. Using code
Optima++, the rate parameters of the San Diego NH3 mechanism (only 21
species and 64 reactions) were optimised against a large collection of
laminar burning velocity data, and concentration data measured in
jet-stirred reactors and burner-stabilised stagnation flame experiments
to develop a compact, yet robust model for CFD simulations. Due to its
small size, the mechanism lacks important chemical pathways, so the
requirement for physically realistic rate coefficients had to be
sacrificed in order to achieve the best possible predictivity for
practical applications. The mechanism has been tested for 70/30 vol%
NH3/H2 mixtures in CFD simulations of a general swirl burner against
experimentally measured concentrations. Its predictions demonstrated
good qualitative and often quantitative agreement with the experimental
data for NO, N2O, and NO2 emissions, and NH3 slip in the whole
equivalence ratio range, while allowing accelerated simulations compared
to other leading mechanisms.
Continue reading...
Reduction-assisted parameter optimization of the ethylene chemistry
in the AramcoMech 2.0 combustion mechanism
Boyang Su, Tibor Nagy, Máté Papp, Tamás TurányiCombust. Flame, 273,
113976 (2025)
Kinetics parameter optimization of the
ethylene chemistry in the AramcoMech 2.0 mechanism (493 species and 2716
reactions) was carried out against a large collection of indirect (1440
data points in 153 data sets) and direct (936 data points in 58 data
sets) experimental data. The indirect data collection consisted of
ignition delay time measurements in shock tubes covering a temperature
range of 930-2230 K and a pressure range of 0.28-63.3 atm, and laminar
burning velocity measurements at preheat temperatures from 298 to 650 K,
and pressures from 0.5 to 10 atm. Due to the large size of the model
and the data collection, direct optimization was not feasible,
therefore, we applied the recently proposed Reduction-Assisted Parameter
Optimization-Based Model Development (RAPOD) procedure. First, using
the Simulation Error Minimization Connectivity Method (SEM-CM), a
reduced mechanism with 75 species and 612 reactions was obtained that
performs similarly to the detailed mechanism regarding the indirect
measurements used. This smaller model could be simulated around 50 times
faster enabling efficient optimization with moderate computational
effort on the large number of experimental targets. Then, influential
reactions of the reduced model were identified using the novel PCALIN
method, which is based on principal component analysis of the local
sensitivity matrix scaled with experimental data uncertainty and
parameter uncertainty. The Arrhenius parameters (ln A, n, E/R) in
18 reactions were optimized within their prior uncertainty domain
against the data collection. Finally, the optimized parameters were
transferred to the original AramcoMech 2.0 mechanism, whose performance
was shown to improve in a similar fashion as that of the reduced model.
The uncertainties of the model results were considerably reduced due to
the significant reduction of the uncertainties of most of the optimized
rate coefficients.
Continue reading...
Identification of well-parameterised reaction steps
in detailed combustion mechanisms –
a case study of ammonia/air flamesAndrás Gy. Szanthoffer, Máté Papp, Tamás Turányi
Fuel, 380,
132938 (2024)
Testing detailed combustion mechanisms typically concludes that some
mechanisms reproduce the experimental data well at most conditions but are inaccurate at other conditions. However, other mechanisms may
perform well under these conditions. A better mechanism (“mosaic mechanism”) may be obtained by identifying the overall best-performing
mechanism and loaning the most important reaction steps and their rate parameters from another mechanism with good performance at the
conditions where the overall best model is ill-performing. A new algorithm based on this approach is presented here, which is
successfully applied using a comprehensive collection of NH3/air laminar burning velocity data (348 data points in 61 data series) and eight recent detailed NH3
combustion mechanisms. The suggested new mosaic mechanism is an improved version of the CEU-2022 mechanism and provides a better
reproduction of the utilised data than the previously published mechanisms. The proposed algorithm can be applied to any chemical
kinetics system and any other types of experiments. All data needed to apply the algorithm to various combustion systems are already available
or can be generated with minimal human effort using the experimental data files, mechanisms, and codes available on the ReSpecTh (https://ReSpecTh.hu) website.
Continue reading...
Modelling of JP-8
distributed combustion using a HyChem mechanism under gas turbine conditionsJanka Borsó, Máté Papp, Viktor Józsa, Tamás Turányi
Results in Engineering,
23, 102596 (2024)
The critical advantages of liquid fuels in aviation
over alternative energy carriers imply their dominance in the upcoming decades. The Mixture Temperature Controlled (MTC) combustion concept
allows spatially homogeneous, efficient burning (distributed combustion) with low pollutant emission. MTC combustion of jet fuel JP-8 was
investigated in an atmospheric burner, and the measured pollutant concentrations in the flue gas were reproduced using the Hybrid
Chemistry (HyChem) approach employing Perfectly Stirred Reactor simulations. Using this robust approach comes with losing spatiotemporal
characteristics if the mixture homogeneity assumption is globally valid. The effect of residence time and pressure under typical gas turbine
operating conditions was investigated. Stable combustion was present above 3 ms residence time at all pressures, while the lower limit was
0.3 ms. The residence time interval of stable operation could be extended by applying flue gas recirculation. NO emission can be reduced
by increasing the operating pressure, while the N
2O
production is dominant only up to 20 bar and in the 10–100 ms residence time range. CO emission vanishes above 10 ms residence time. Ultra-low
emission operation requires >20 bar pressure and 10 ms residence time with distributed combustion, requiring larger combustion chambers for
future jet engines.
Optimization of a methanol/NOx combustion mechanism based on a large amount of experimental dataMárton Kovács, Máté Papp, András Gy. Szanthoffer, István Gy. Zsély, Tibor Nagy, Tamás Turányi
Fuel, 375,
132544 (2024)
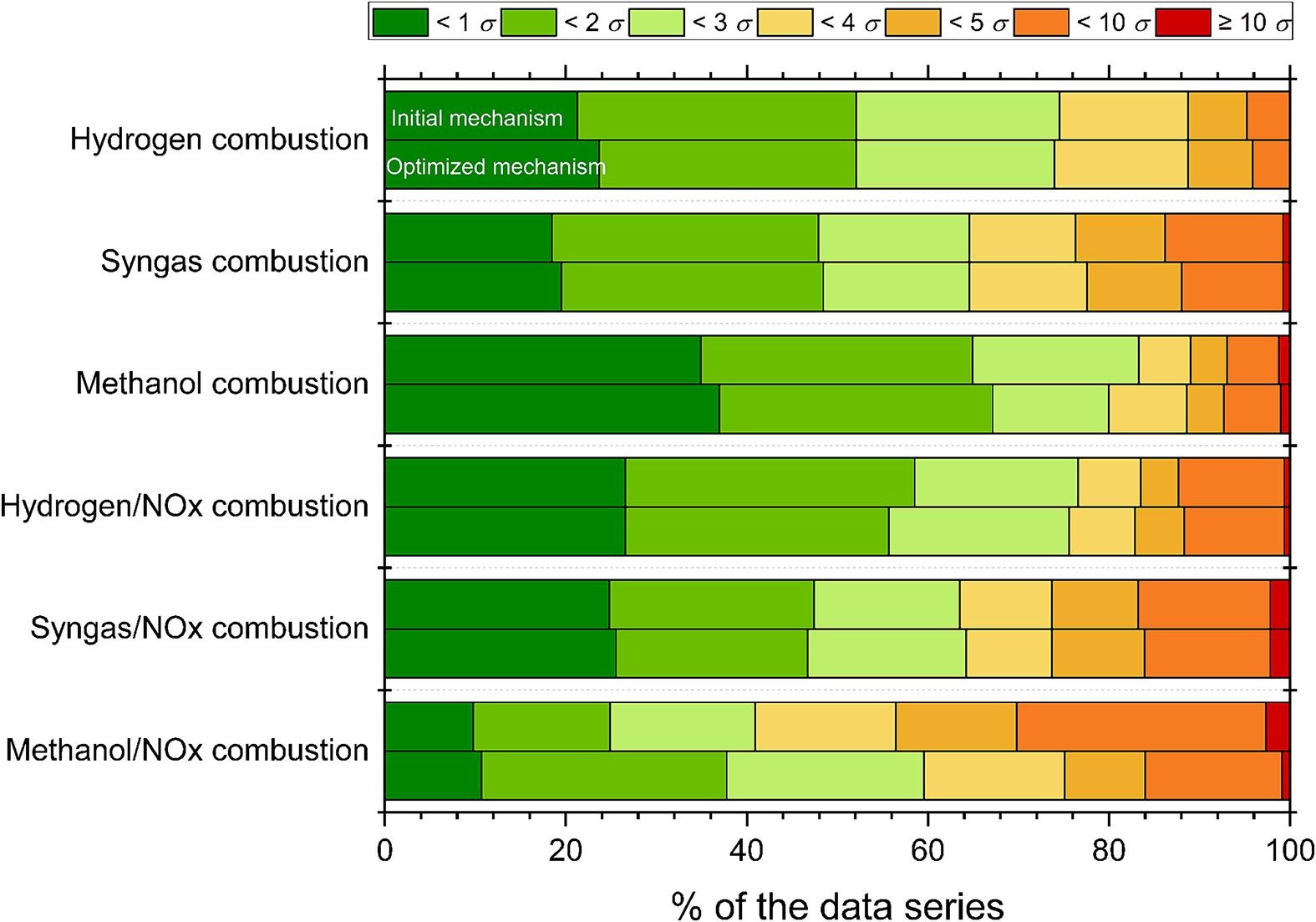
Investigating
the methanol (CH3OH) / nitrogen-oxides (NOx) combustion system is an
important task since methanol is a promising alternative to fossil
fuels, and its interactions with nitrogen oxides are significant due to
environmental effects. The performances of the recently available
detailed mechanisms in simulating the experimental results are still
unsatisfactory. The aim of this work is to develop a more reliable
reaction mechanism using parameter optimization. First, the
Glarborg-2018 mechanism was updated with the rate parameters of the
previously optimized H2/NOx and methanol mechanisms of ELTE. A large
collection of literature data was compiled, which consists of direct
measurements and theoretical determinations of the rate coefficients
(2175 data points in 130 data series), indirect measurements of the
formaldehyde (CH2O) /NOx and CH3OH/NOx system in homogenous reactors
(2373 data points in 225 data series), and the neat CH3OH and CH2O
subsystems in homogenous reactors and flames (689 data points in 68 data
series). Using code Optima++, we optimized the rate parameters of the
24 most important elementary reactions that were identified by the
recent PCALIN (Principal Component Analysis of the Parameter-Uncertainty
and Data-Uncertainty Scaled Local Sensitivity Matrix with Linear
Corrections) active parameter selection method as most influential. The
optimized rate coefficients were assessed in detail and compared with
literature data. The optimized mechanism can reproduce the CH2O/NOx and
CH3OH/NOx combustion experimental data on average within their 3.5σ
experimental uncertainty, which means it performs significantly better
than the previously published mechanisms, which have average errors
larger than 5σ. The reproduction of neat CH3OH and CH2O experimental
data also improved. The optimized mechanism was also tested on
experimental data of the H2, H2/NOx, syngas, and syngas/NOx combustion
systems. In all cases, the optimized mechanism reproduced these
experimental data better than the initial mechanism, although these data
were not used as optimization targets.
Continue reading...
Mechanism development for larger alkanes by autogeneration and rate rule optimization:
the case study of pentane isomersPengzhi Wang, Sirio Brunialti, Máté Papp, S. Mani Sarathy, Tamás Turányi, Henry J. Curran, Tibor Nagy
Proc. Combust. Inst.,
40, 105408 (2024)
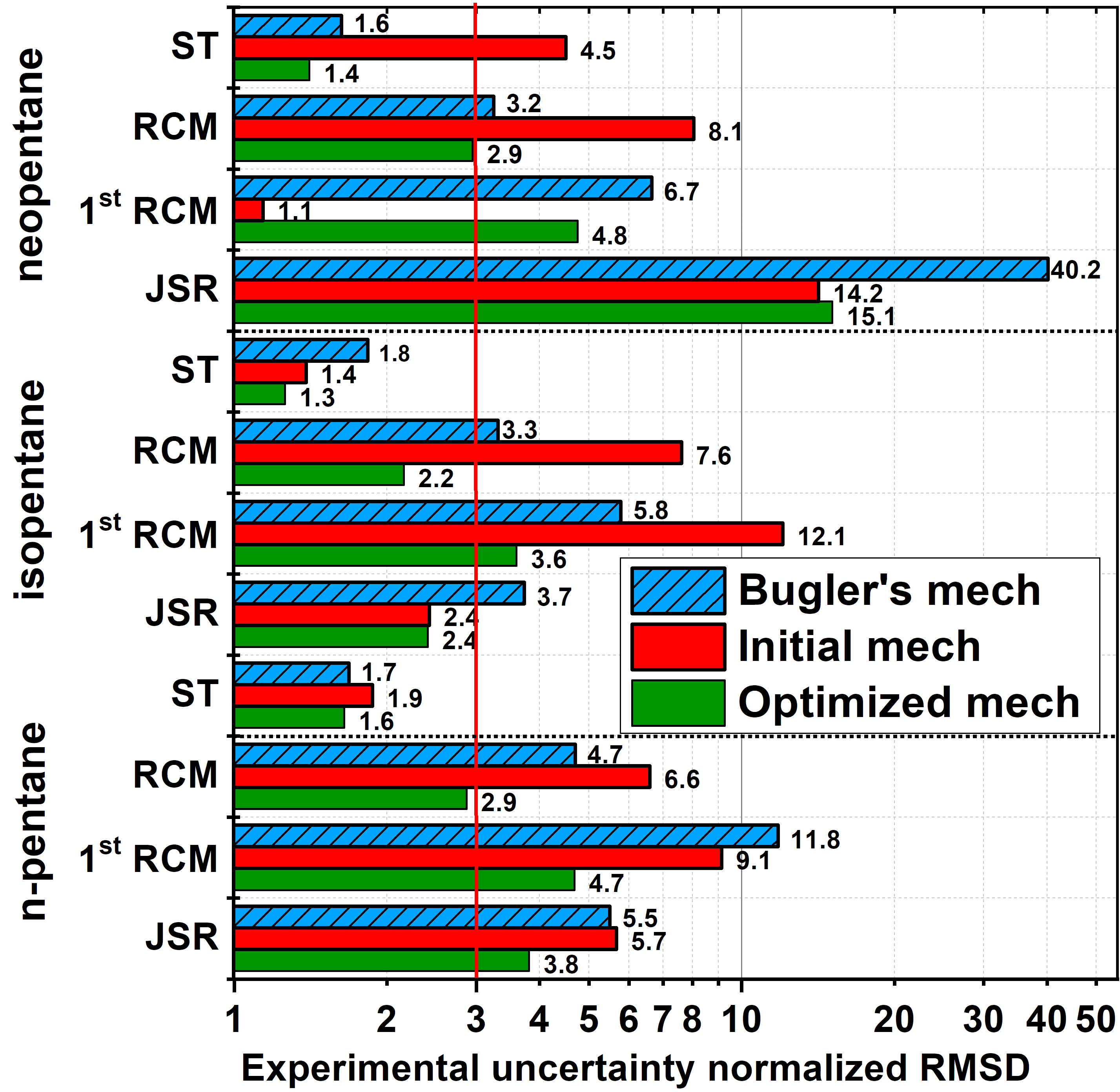
The
core chemistry and thermodynamic data of large alkanes in the NUIGMech
mechanism were recently updated. In the present work, the set of rate
rules for large alkanes is optimized against experimental data to
improve the predictivity of the mechanism. As a starting step of
developing a consistent set of rate rules for any larger alkane, we
optimized the mechanism of pentane isomers, whose mechanism was
generated based on 185 rate rules in 24 reaction classes using code
MAMOX++. Including the core chemistry, the mechanism contained 1427
species and 6676 reactions. For the efficient optimization of such a
large mechanism, the Optima++ code was extended to rate rules and was
linked with the Zero-RK simulation code. As reference data, first-stage
and total ignition delay times measured in shock tubes and rapid
compression machines, and species concentrations measured in jet-stirred
reactors were collected in wide ranges of conditions. The prior
uncertainties of the Arrhenius equations of the 185 rate rules were
determined based on a review of alkanes’ rate constant studies. The
PCA-SUE method was used for the selection of the influential rate rules.
This method identified 94 important rate rules, whose Arrhenius
parameters were subsequently optimized within their prior uncertainty
ranges using Optima++ against a representative subset of the data
collection with a moderate computational effort. The optimization
significantly improved the accuracy of the mechanism, which now performs
significantly better even than the Bugler et al. mechanism (PROCI,
2017). The present study has demonstrated the effectiveness of the
proposed methodology, thereby paving the way to the optimization of a
complete set of rate rules that can be used for the generation of a
reliable combustion mechanism for any larger alkane, and with some
extensions even for unsaturated fuels or oxygenated fuels such as
biodiesels.
Ammonia production by microbubbles: a theoretical analysis of achievable energy intensityFerenc Kubicsek, Áron Kozák, Tamás Turányi, István Gyula Zsély, Máté Papp, Al-Awamleh Ahmad, Ferenc Hegedűs
Ultrasonics Sonochemistry, 106, 106876 (2024)
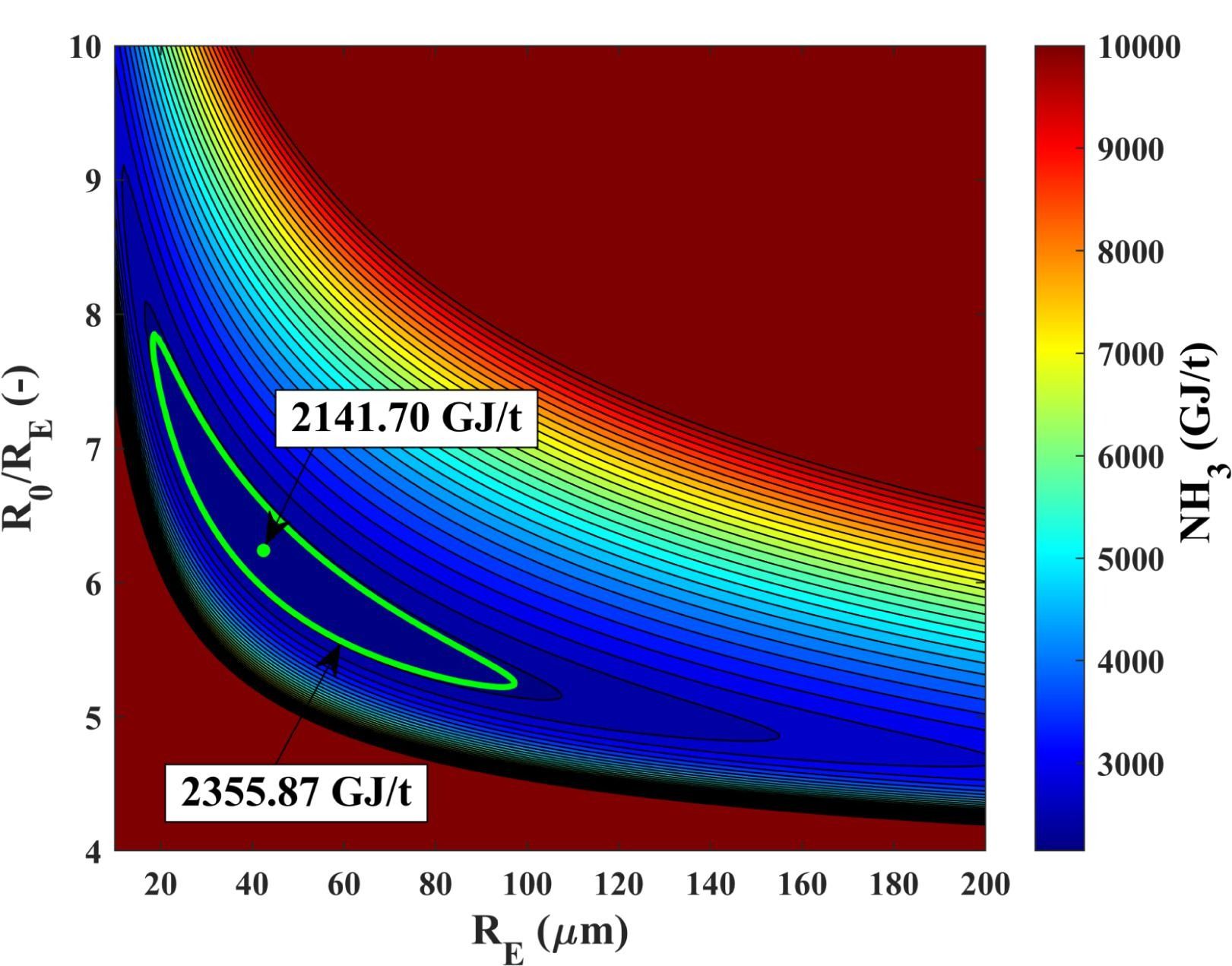
The
present paper studies the energy intensity of ammonia production by a
freely oscillating microbubble placed in an infinite domain of liquid.
The initial content of the bubble is a mixture of hydrogen and nitrogen.
The bubble is expanded isothermically to a maximum radius, then it is
“released” and oscillates freely. The input energy is composed of the
potential energy of the bubble at the maximum radius, the energy
required to produce hydrogen, and the pumping work in case a vacuum is
employed. The chemical yield is computed by solving the underlying
governing equations: the Keller–Miksis equation for the radial dynamics,
the first law of thermodynamics for the internal temperature and the
reaction mechanism for the evolution of the concentration of the
chemical species. The control parameters during the simulations are the
equilibrium bubble size, initial expansion ratio, ambient pressure, the
initial concentration ratio of hydrogen and the material properties of
the liquid. At the optimal parameter setup, the energy intensity is that
is times higher than the best available technology, the Haber–Bosch
process. In both cases, the hydrogen is generated via water
electrolysis.
Dependence of ignition delay
time on its definition −
A case study on methane ignitionBoyang Su, Máté Papp, Peng Zhang, Tamás Turányi
Combust. Flame,
262, 113364 (2024)
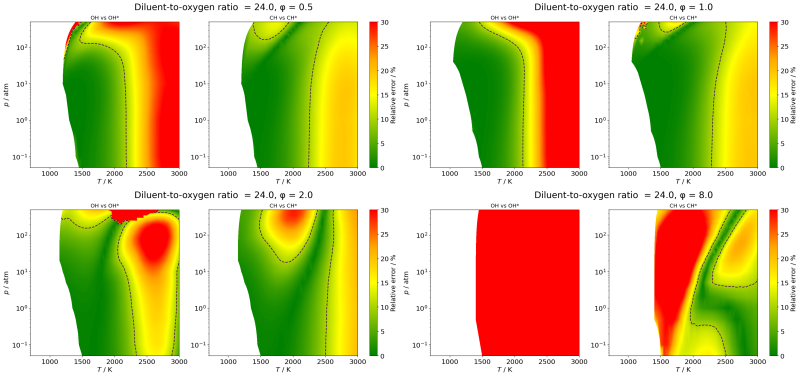
Ignition delay time (IDT, τ)
is the time period from the onset of
a given temperature and pressure of a combustible gas mixture to
the time of ignition. The time point of ignition is defined as
reaching a given
condition of pressure, temperature, or one of the species
concentrations. In measurements, ignition of hydrocarbons and
oxygenates is usually
detected based on monitoring the change of pressure or the
concentrations of species OH*, CH*, or CO2. In simulations, ignition is also
detected based on the temperature profile. Simulated ignition of CH4/O2/N2
mixtures was investigated in a wide range of initial temperature
(700-3000 K), pressure (0.05-500 atm), and equivalence ratio (φ=0.03-8.0)
at
two diluent-to-oxygen ratios: 3.76 (corresponding to air), 24.0
(corresponding to the conditions of many shock tube measurements).
The
agreement between τp and τOH* was
good below initial temperature 2500 K and 0.5<φ<2.0, while
the agreement between τp and values of τCH*,
τCO2 and τT was much more than 10%
for most of the conditions. The τOH vs. τOH*
agreement is poor above φ=3.0 for most conditions. The τCH
vs. τCH* agreement is good in the range of φ
=
0.4 to 1.5, below 2200 K and 100 atm. The experimental
determinations of IDTs should consistently be reproduced by
simulations using exactly the
same IDT definition. When different IDT definitions are used in
various measurements or simulations, the τ values are expected to be in
good agreement only in restricted ranges of conditions.
Continue reading...
Comparison of the performance of ethylene combustion mechanisms
Boyang Su, Máté Papp, István Gy. Zsély, Tibor Nagy, Peng Zhang, Tamás Turányi
Combustion and Flame, 260, 113201 (2024)
Ethylene is a key intermediate in the combustion and pyrolysis
of larger hydrocarbons. A large set of literature experimental data covering wide ranges of conditions on ethylene combustion was collected:
ignition delay times measured in shock tubes (1160 data points in 114 data series) and rapid compression
machines (83/5); concentration measurements carried out in jet-stirred reactors (1586/157) and flow reactors (649/59); and laminar burning
velocities (882/59). 14 detailed reaction mechanisms were investigated using the collected experimental data. The results show significant
differences between the performances of the various mechanisms. The four best-performing mechanisms are Aramco-2.0-2016, Aramco-3.0-2018,
Yang-2020 and NUIGMech-1.1-2020 based on all included experimental data. The best-performing model Aramco-2.0-2016 was further investigated by
local sensitivity analysis. Several reaction steps were found to be important for simulating the experiments: many reactions of the
hydrogen, syngas and C1 hydrocarbons oxidation systems, furthermore reactions C2H3 + O2 = CH2CHO + O and C2H4 + OH = C2H3 + H2O which are specific for ethylene combustion.
Effect of the variation of oxygen concentration on the laminar burning velocities of hydrogen-enriched
methane flames
Sven Eckart, István Gyula Zsély, Hartmut
Krause, Tamás Turányi
International Journal
of Hydrogen Energy, 49, 533-546 (2024)
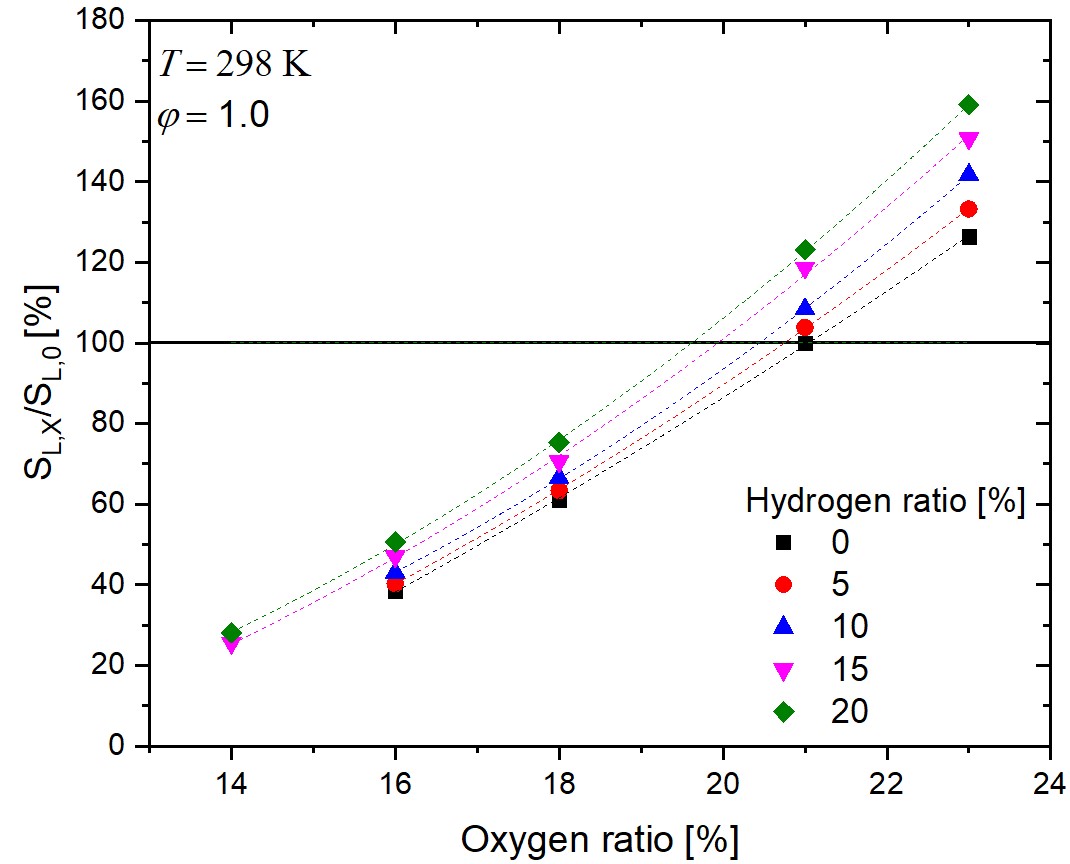
The
combustion properties of hydrogen-containing fuel mixtures and the
effect of the variation of the oxygen content of the oxidizer are
at the center of recent research interest. Laminar burning velocity
measurements
with varied oxygen content can help in the validation of reaction
mechanisms for better simulations of combustion systems using
exhaust gas
recirculation (EGR) or oxygen-enriched atmosphere. Such
measurements were carried out in hydrogen-enriched methane-air
flames using the heat flux
method with higher accuracy and a wider range of initial oxygen
and hydrogen concentrations compared to the similar studies in the
literature.
The mole fractions of the hydrogen and oxygen contents of the
initial fuel and oxidizer mixtures were varied between xH2 = 0 and 0.20, and
xO2 = 0.14 and 0.23, respectively. The initial gas
temperature
and pressure were 298 K and 1 bar, respectively. It is
demonstrated that the increase of combustion rate by the hydrogen
enrichment can be
compensated with the decrease of the oxygen content. This
compensating effect was investigated in detail in a wide range of
equivalence ratio
(φ). The experimental data were simulated with 11 widely
used methane combustion reaction mechanisms. The prediction
accuracies of the
mechanisms at lean and rich equivalence ratios were significantly
different and the important reaction steps were identified using
sensitivity analysis for three mechanisms. Mechanisms POLIMI-2014
and Caltech-2015 gave the best overall predictions.
Continue reading...
Reaction kinetics of hydrogen combustion
Tamás Turányi
Chapter 2 in book: Hydrogen for future thermal engines, (ed: Efstathios - Alexandros Tingas), Springer Nature, 2023

The
reaction kinetics of hydrogen combustion was intensively studied
already in the first half of the 20th century. The first, second and
third explosion limits were discovered, and mechanistic explanations
were given. However, fine details of the reaction mechanism and the
exact rate parameters are still not known. New reaction steps were
proposed even in the last few years. The explosive—non-explosive regions
are separated by an inverted S-shape curve in the
T −
p
plane. The backbone of the curve is approximately equal to the line
where the rates of reactions H + O2 → OH + O and H + O2 + M → HO2 + M
are equal. The regions of strong and weak explosions are below and above
the backbone line, and these regions are dominated by H/O/OH and
HO2/H2O2 catalytic cycles, respectively. In a small vessel, the
adsorption of radicals HO2 and H form the upper and lower branches of
the curve, respectively. All qualitative features of the hydrogen
combustion system can be explained by a 10-step mechanism. Several web
sites are recommended here which contain comprehensive collections of
recent hydrogen combustion mechanisms, direct and indirect experimental
data, and theoretical determinations.
András Szanthoffer, István Gyula Zsély, László Kawka, Máté Papp, Tamás Turányi
Applications in Energy and Combustion Science (AECS), 14, 100127 (2023)
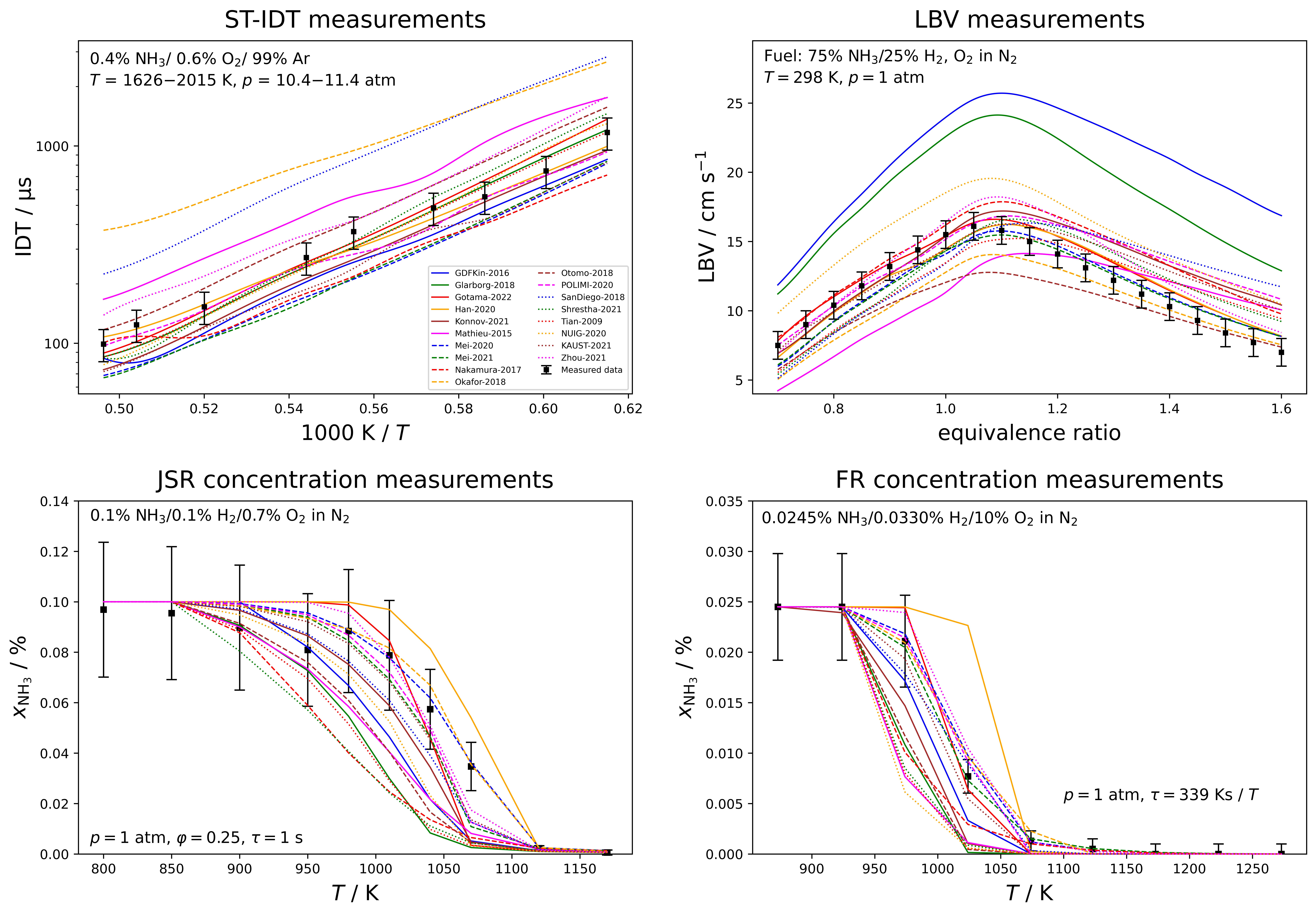
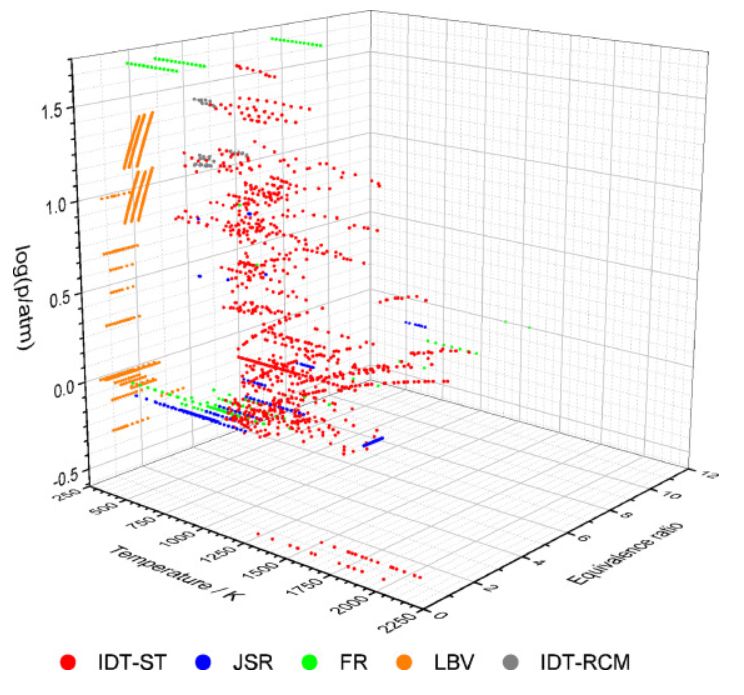
A possible solution to improve the combustion properties of ammonia is
to blend it with other fuels. Two of the most usually used co‑fuels are hydrogen and syngas (H2/CO). To investigate the chemistry of
the co-combustion with these fuels, a large amount of indirect experimental data for the combustion of neat NH3, and NH3/H2
and NH3/syngas fuel mixtures were collected from the literature
including ignition delay times measured in shock tubes, concentration measurements in jet stirred and flow reactors, and laminar burning
velocity measurements. Altogether, 4898 data points (in 472 data series) were recorded which cover wide ranges of equivalence ratio, temperature,
and pressure. These experimental data are available in data files in the ReSpecTh site (http://respecth.hu). The
performances of 18 recently published detailed reaction mechanisms were quantitatively assessed using the collected experiments. There are
significant differences between the performances of the models, and the performance of a mechanism may also vary significantly with the different
types of experiments. The best‑performing mechanisms are POLIMI‑2020, KAUST‑2021, and Otomo‑2018 for NH3/H2 fuel mixtures,
and Shrestha‑2021, Mei‑2021, and Mei‑2020 for NH3/syngas
systems. The results indicate that further mechanism development is needed to reproduce the measurements more accurately. Local sensitivity analysis
was carried out on the kinetic and thermodynamic parameters of the best‑performing mechanisms. Even though the investigated models have
different parameter sets, the most important reactions and thermodynamic properties are similar. The most important reactions are not the same for
the different types of experiments but most of them include the NH3,
NH2, and/or NNH species. Among the thermodynamic parameters,
model outputs are most sensitive to the data of NH3 and NH2.
Continue reading...
Efficient numerical methods for the optimization of large kinetic reaction mechanisms
Simret Kidane Goitom, Máté Papp, Márton Kovács, Tibor Nagy, István Gy. Zsély, Tamás Turányi, László Pál
Combustion Theory Modelling, 26, 1071-1097 (2022)
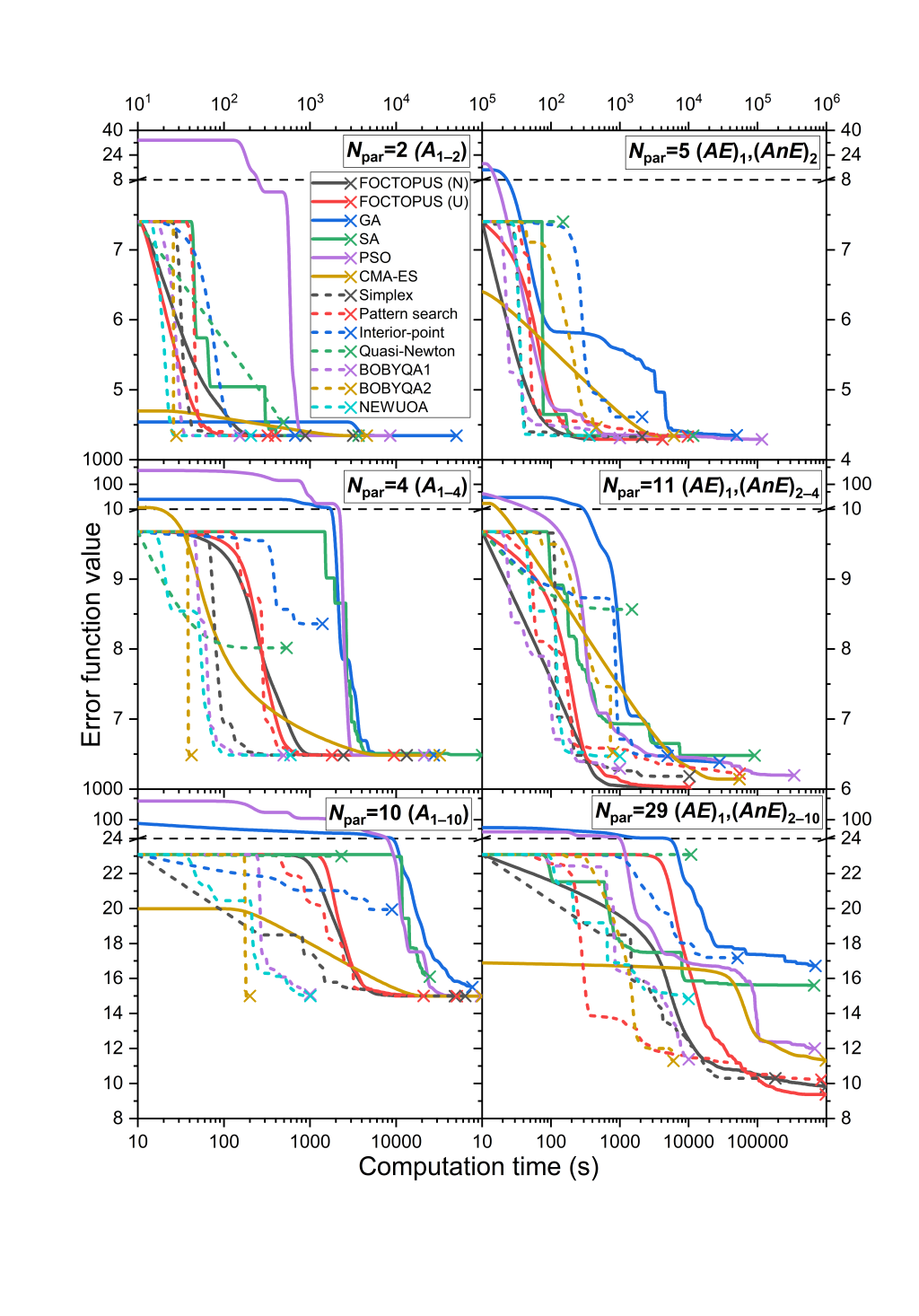
Optimization of detailed combustion mechanisms means that the corresponding kinetic model is fitted to experimental data via optimizing their important rate and thermodynamic parameters within their domain of uncertainty. Typically, several dozen parameters are fitted to several hundred to several thousand data points. Many numerical optimization methods have been used, but the efficiency of these methods has not been compared systematically. In this work, parameters of a H2/O2/NOx mechanism (214 reaction steps of 35 species) were fitted to 1552 indirect (ignition delay times measured in shock tubes and concentrations measured in flow reactors) and 755 direct measurements. Three test cases were investigated: (1) fitting the Arrhenius parameters of 2 reaction steps to 732 data points; (2) fitting the Arrhenius parameters of 4 reaction steps to 1077 data points; (3) fitting the Arrhenius parameters of 10 reaction steps to 2307 data points. All three cases were investigated in two ways: fitting the A-parameters only and fitting all Arrhenius parameters (5, 11 and 29 parameters, respectively). A series of global (FOCTOPUS, genetic algorithm, simulated annealing, particle swarm optimization, covariance matrix adaptation evolutionary strategy (CMA-ES)) and local (simplex, pattern search, interior-point, quasi-Newton, BOBYQA, NEWUOA) optimization methods were tested on these cases, some of them in two variants. The methods were compared in terms of the final error function value and number of error function evaluations. The main conclusions are that the FOCTOPUS resulted in the lowest final error value in all cases, but this method required relatively many error function evaluations. As the task became more difficult, more and more methods failed. A variant of the BOBYQA method looked stable and efficient in all cases.
Identification of homogeneous chemical kinetic regimes of methane-air ignitionÉva Valkó, Máté Papp, Peng Zhang, Tamás Turányi
Proc. Combust. Inst., 39, available online (2023)
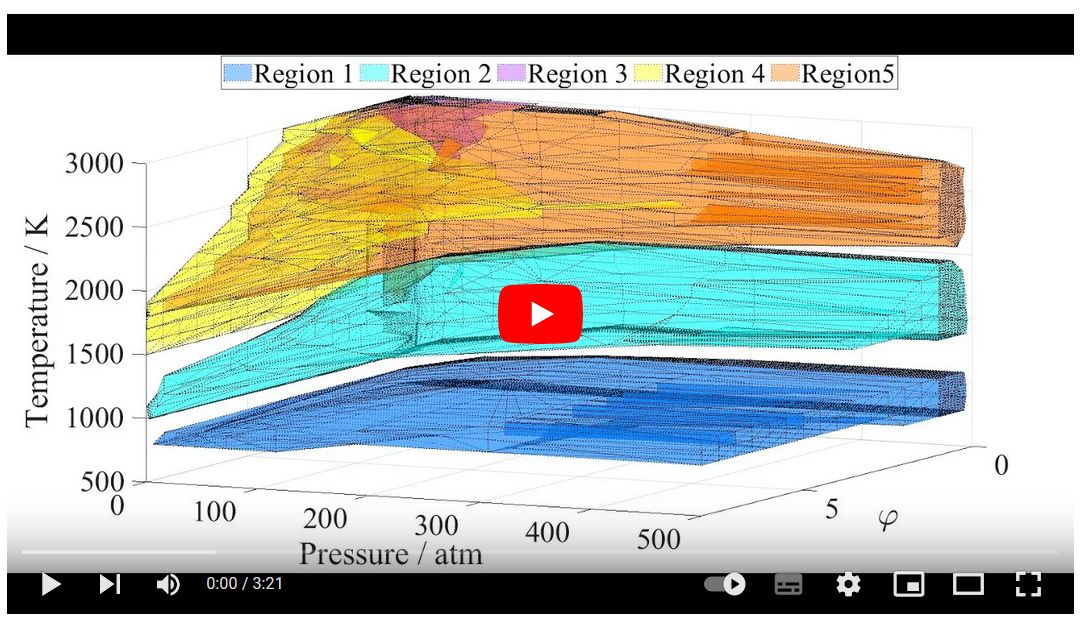
Sensitivity
analysis results for ignition delay time (IDT) may be very different
depending on the initial temperature, pressure and equivalence ratio φ,
but similar in some regions of these variables. This phenomenon was
investigated systematically by carrying out ignition simulations and
local sensitivity calculations of methane−air mixtures using the
Aramco-II-2016 mechanism at 14,417 combinations of initial temperature
(changed between 500 and 3000 K), initial pressure (0.05−500 atm) and φ
(0.05−8.0) values. The cluster analysis of the sensitivity vectors
identified five large kinetically homogeneous regions. Each region has
well defined borders in the (T, p φ) space and can be characterized by
different sets of important reactions. The related kinetic scheme is
very different in each region. Regions 1 and 2 are dominated by
catalytic cycles based on species CH3O2/CH3O2H and HO2/H2O2/CH3O,
respectively. In regions 3, 4, and 5 the H atoms are converted to CH3 in
an identical chain branching sequence, but the back conversion is via
three different routes. Literature experimental data on the IDTs of
methane−air mixtures were sorted according to these five regions.
Regions 1 to 5 contain 214, 328, 3, 0, and 237 experimental data points,
respectively. In regions 1, 2 and 5 the data points are well reproduced
by the Aramco-II-2016 mechanism, but little or no experimental
information is available about kinetic regions 3 and 4. Further
experimental exploration of the ignition of methane−air mixtures may aim
the study of these regions. A similar approach can be used for the
characterization of other combustion systems and sorting the related
experimental data.
A novel active parameter selection strategy for the efficient optimization of combustion mechanismsMárton Kovács, Máté Papp, Tamás Turányi, Tibor Nagy
Proc. Combust. Inst., 39, available online (2023)
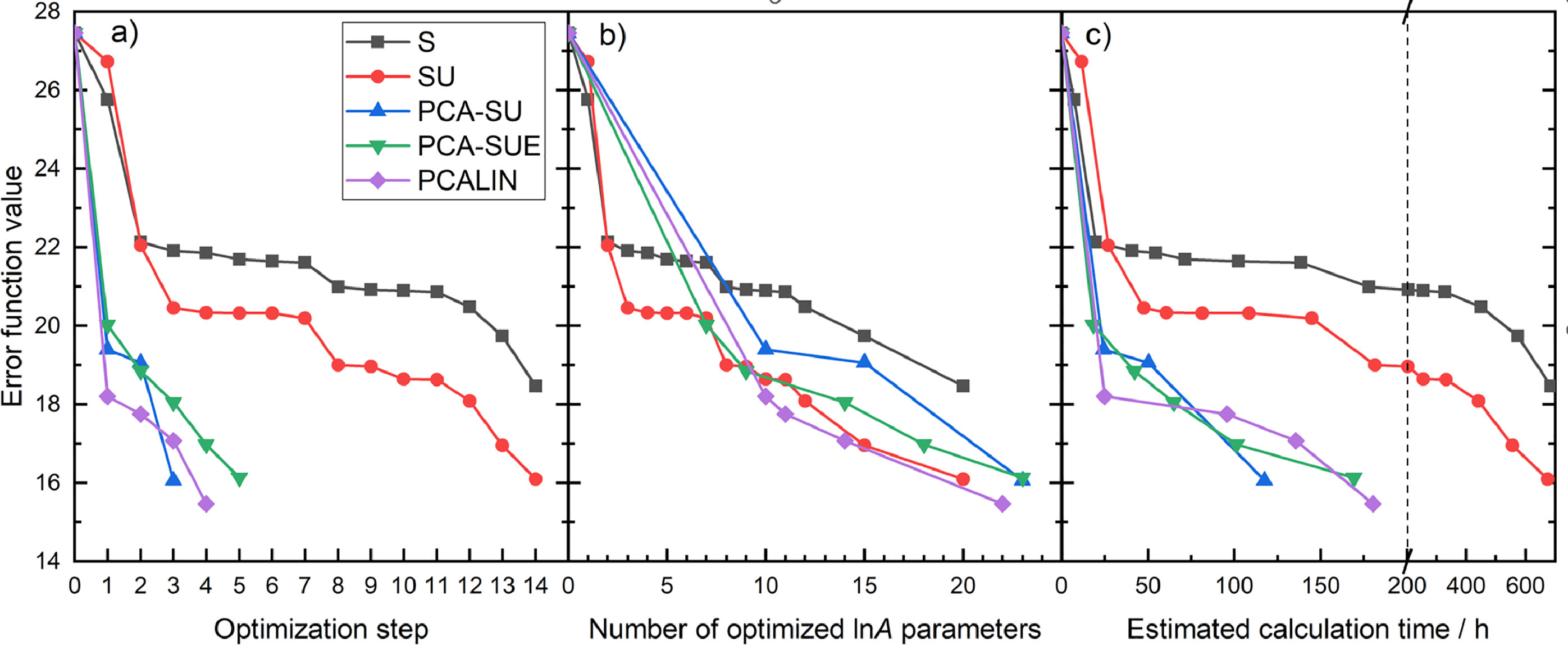
Optimization
of large combustion mechanisms means that a few dozen parameters
(called active parameters) are optimized within their uncertainty limits
to achieve a better reproduction of the experimental data, which is
usually measured by a mean square error function. In previous studies,
the active parameters were selected based either on their local
sensitivity coefficients (strategy S) or on the products of local
sensitivity coefficient and a corresponding uncertainty parameter
(strategy SU). This latter measure is known by various names:
optimization potential, sensitivity-uncertainty index or
uncertainty-weighted sensitivity coefficient. In this work, we proposed
three novel active parameter selection strategies of increasing
complexity (PCA-SU, PCA-SUE, PCALIN) and demonstrated their superior
performance in the optimization of pre-exponential factors (A) in a
methanol/NOx combustion mechanism (562 reaction steps of 70 species)
against 2360 data measured in shock tube, JSR and flow reactor
experiments. The novel methods are based on the principal component
analysis (PCA) of sensitivity matrices scaled by the uncertainties of
parameters (U) and the uncertainty of the experimental data (E). These
PCA-based methods take into account parameter correlations and designate
parameter groups and corresponding relevant subsets of experimental
data, thereby a factor of 4–7 savings in optimization time was achieved
over the S and SU methods. PCA-SUE method performed better than the
PCA-SU as it also considered the uncertainty of the experimental data.
The PCALIN strategy is similar to PCA-SUE, but it also considers the
linear change (LIN) of the error function, which depends on the
simulation error of experimental data, and thereby it could provide the
most accurate models as a function of the number of active parameters.
Based on the PCALIN strategy, fitting all three Arrhenius parameters
resulted in further improvements, however, it provided moderate
improvements over simple A-factor tuning and required significantly more
computer time.
Comparison and analysis of butanol combustion mechanisms
Martin Bolla, Máté Papp, Carsten Olm, Hannes Böttler, Tibor Nagy, István Gy. Zsély, Tamás Turányi
Energy&Fuels, 36, 11154–11176 (2022)
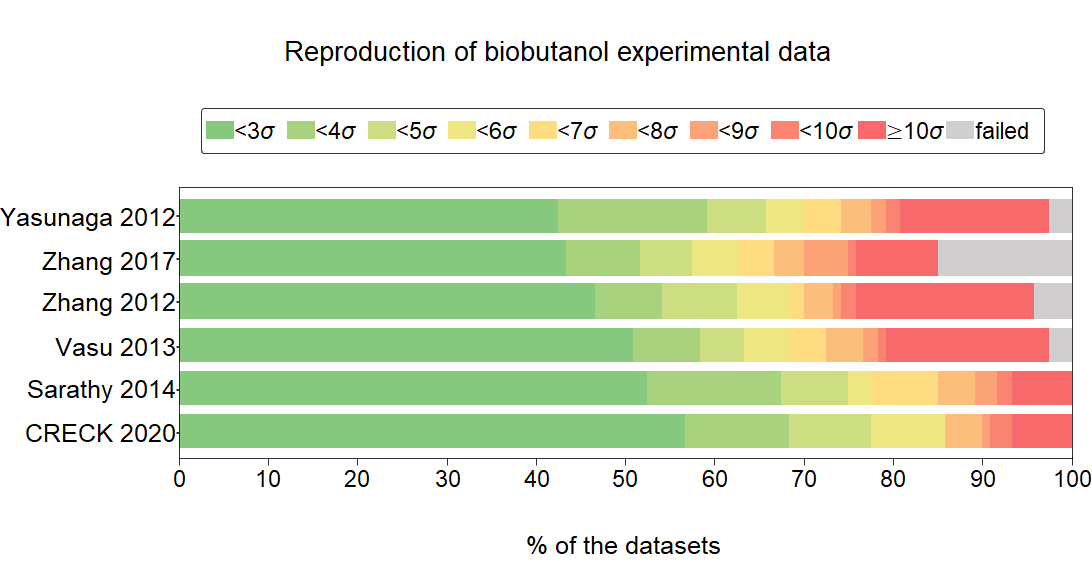
A
detailed review of the performances of 24 butanol combustion
mechanisms, published between 2008 and 2020, is given using a
comprehensive experimental data collection (89,388 data points in 266
datasets from 32 publications). The data cover wide ranges of
equivalence ratio (φ = 0.38–2.67), diluent ratio (0.15–0.98), initial
temperature (672–1886 K), and pressure (0.9–90 atm). The collection
includes ignition delay time measurements in shock tubes and rapid
compression machines, concentration determinations in shock tubes,
jet-stirred reactors, flow reactors, and laminar burning velocity
measurements. The experimental data were recorded in ReSpecTh Kinetics
Data Format (RKD format) v.2.3 XML data files, which are available in
the ReSpecTh site (http://respecth.hu). The standard deviations of the
measurements were estimated using both the published experimental
uncertainty and the scatter error of the datasets determined by code
Minimal Spline Fit. Mechanism CRECK 2020 was found to be the best
mechanism for n-butanol (biobutanol) combustion, while the mechanisms
Sarathy 2014, Vasu 2013, and Yasunaga 2012 (in this order) were the best
considering the experimental data for all isomers. A part of the
simulations failed, and to improve the ratio of successful simulations,
the code ThermCheck was created, which detects discontinuities and
nonsmoothness of thermodynamic functions defined by NASA polynomials
provided with the published mechanisms and corrects them by tuning their
coefficients. Local sensitivity analysis applied to the experimental
conditions was used to identify the most important reaction steps of the
mechanism Sarathy 2014. The sensitivity analysis was extended to the
adiabatic ignition of n-butanol–air mixtures by systematically changing
the initial temperature and pressure. All butanol combustion mechanisms
were also tested on a hydrogen combustion data collection, which
indicated that some of them were inaccurate due to their inadequate
hydrogen combustion reaction block. Suggestions were given for the
improvement of the Sarathy 2014 mechanism.
The importance of chemical mechanisms in sonochemical modelling
Cs. Kalmár, T. Turányi, I. Gy. Zsély, M. Papp, F. Hegedűs
Ultrasonics Sonochemistry, 83, 105925 (2022)
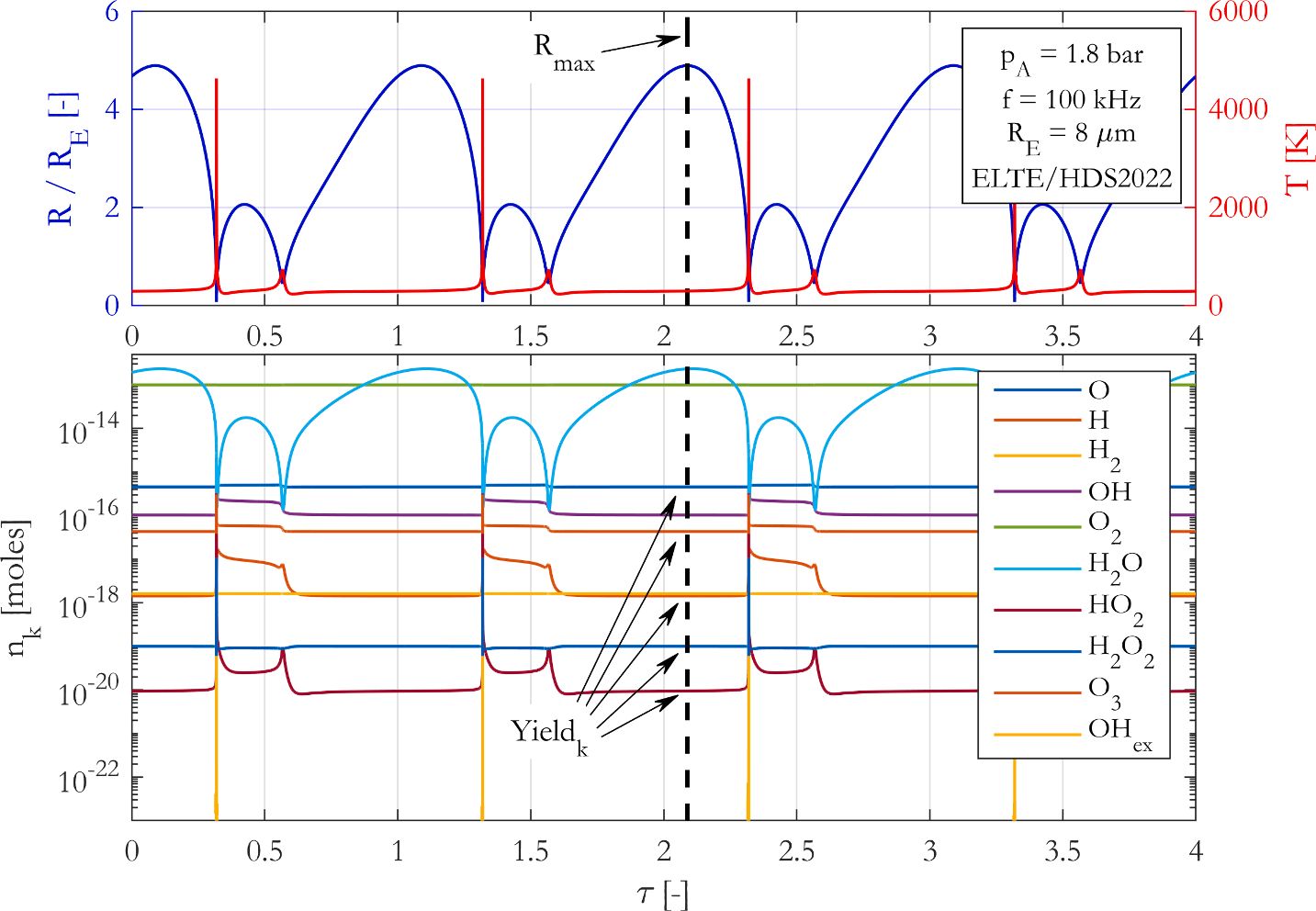
A
state-of-the-art chemical mechanism is introduced to properly describe
chemical processes inside a harmonically excited spherical bubble placed
in water and saturated with oxygen. The model uses up-to-date
Arrhenius-constants, collision efficiency factors and takes into account
the pressure-dependency of the reactions. Duplicated reactions are also
applied, and the backward reactions rates are calculated via suitable
thermodynamic equilibrium conditions. Our proposed reaction mechanism is
compared to three other chemical models that are widely applied in
sonochemistry and lack most of the aforementioned modelling issues. In
the governing equations, only the reaction mechanisms are compared, all
other parts of the models are identical. The chemical yields obtained by
the different modelling techniques are taken at the maximum expansion
of the bubble. A brief parameter study is made with different pressure
amplitudes and driving frequencies at two equilibrium bubble sizes. The
results show that due to the deficiencies of the former reaction
mechanisms employed in the sonochemical literature, several orders of
magnitude differences of the chemical yields can be observed. In
addition, the trends along a control parameter can also have dissimilar
characteristics that might lead to false optimal operating conditions.
Consequently, an up-to-date and accurate chemical model is crucial to
make qualitatively and quantitatively correct conclusions in
sonochemistry.
Comparison of methane combustion mechanisms using laminar burning velocity measurements
Peng Zhang, István Gy. Zsély, Máté Papp, Tibor Nagy,
Tamás Turányi
Combust Flame, 238, 111867 (2022)
Publication Date: December 22, 2021
Large
amount of experimental data for laminar burning velocity (LBV)
measurements of methane (+H2/CO) − oxygen − diluent mixtures (5500 data
points in 646 datasets) covering wide ranges of equivalence ratio,
diluent ratio, cold side temperature and pressure were collected from
111 publications. The diluents included N2 , H2O, CO2 , Ar and He. The
data files are available on the ReSpecTh site (http://respecth.hu).
Performances of 12 methane combustion mechanisms on reproducing these
LBV
measurements were analyzed according to experiment types and
conditions. Most mechanisms could predict well the LBVs for
stoichiometric and fuel-lean mixtures and for diluent ratios higher than
60%. The performances of several mechanisms were relatively poor at
other conditions. Focusing on the operating conditions of natural gas
engines, we recommend the application of mechanisms FFCM-I-2016,
SanDiego-2014, and NUIG1.1-2021 for engine simulations. Mechanisms
Aramco-II-2016, Konnov-2009, Caltech-2015 and Glarborg-2018 have the
lowest average errors for the reproduction of all available methane LBV
data.
Using local sensitivity analysis on the most accurate
mechanisms, we identified 29 important elementary reactions, which,
however, were not present in all the 12 mechanisms. We also collected
large amount of directly measured and theoretically calculated rate
coefficients for these reactions and compared them with the rate
coefficients used in the 12 mechanisms. Reactions found important in any
of the Aramco-II-2016, Konnov-2009 and Glarborg-2018 mechanisms, but
missing from the Aramco-II-2016, Konnov-2009, Glarborg-2018,
Caltech-2015, FFCM-I-2016 and NUIG1.1-2021 mechanisms were added to
these six mech-
anisms to investigate if the extended mechanism
performs better than the original one. Some of the extended mechanisms
became the best performing mechanisms.
Comparison of Methane Combustion Mechanisms Using Shock Tube and Rapid Compression Machine Ignition Delay Time MeasurementsP. Zhang, I. Gy. Zsély, V. Samu, T. Nagy, T. Turányi
Energy Fuels,
35, 12329–12351 (2021)
Publication Date: July 9, 2021
https://doi.org/10.1021/acs.energyfuels.0c04277-
We intended to collect all published experimental data on the ignition methane - oxygen mixtures, with possible added fuel: H2 and CO; possible diluents: N2, Ar, He). These included shock tube and RCM data (5521 data points in 643 datasets from 76 publications), covering wide ranges of temperature T, pressure p, equivalence ratio φ, and diluent concentration. Thirteen recent methane combustion mechanisms were tested against these experimental data.
Design of combustion experiments using differential entropyÉva Valkó, Máté Papp, Márton Kovács, Tamás Varga,
István Gy. Zsély, Tibor Nagy, Tamás Turányi
Combustion Theory and Modelling,
26, 67-90 (2022)
Publication Date: November 9, 2021
https://doi.org/10.1080/13647830.2021.1992506-
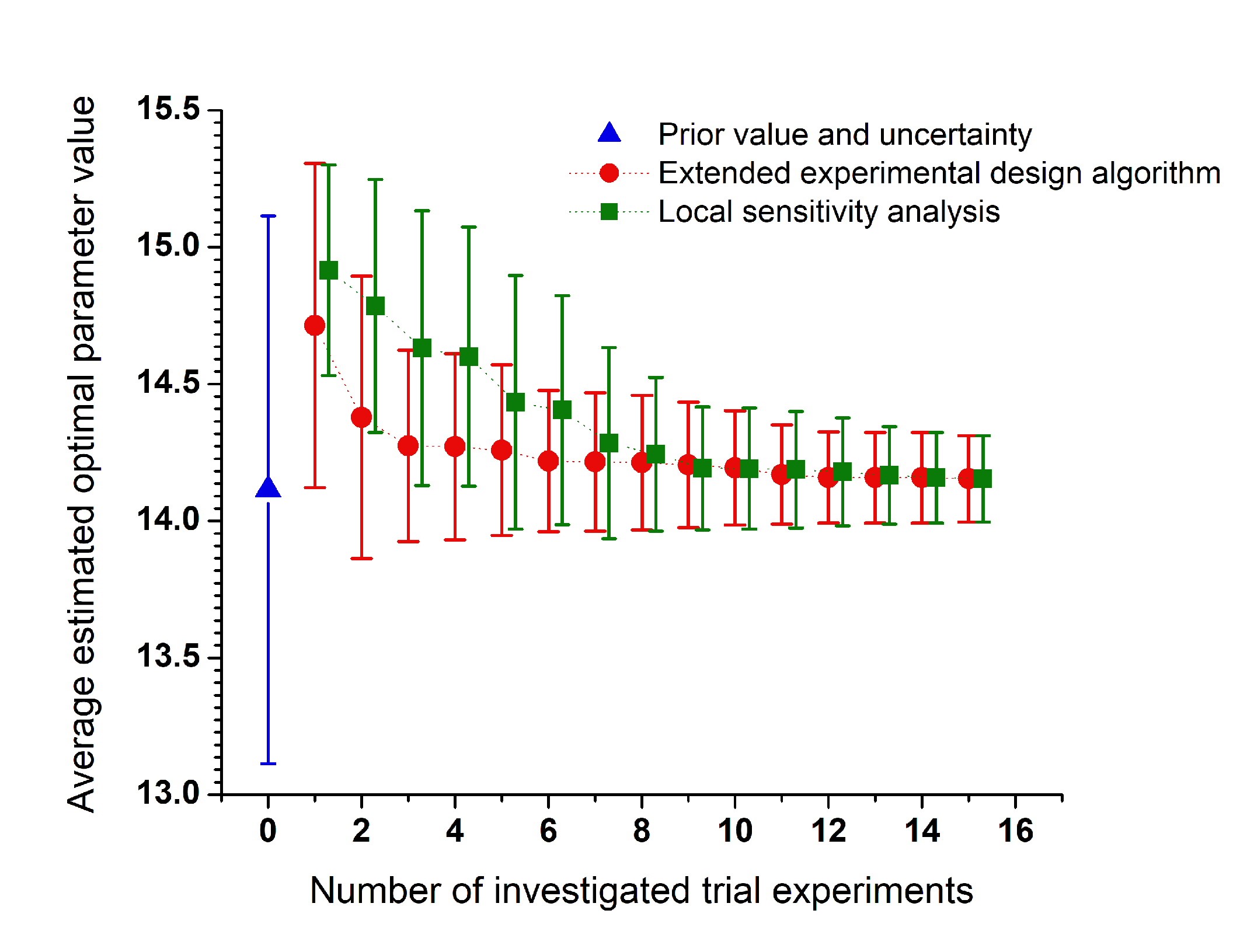
The
aim of several combustion experiments is the determination of the rate
coefficients of important elementary reactions. Sheen and Manion (J.
Phys. Chem. A, 118 (2014) 4929–4941) suggested a method for the design
of shock tube experiments based on differential entropy. Their method
was modified and extended in this work. In the extended method, both the
experimental and residual errors of the measurements are considered at
the calculation of the posterior uncertainty of the determined rate
parameters, the differential entropy matrix is calculated in an
analytical way and the net information flux value is calculated for each
suggested experimental point. In an iterative procedure, all
investigated experimental points with negative net information flux
values are discarded and the remaining experimental conditions are
recommended for the measurements. The most valuable candidate
experimental points can be determined based on the net information flux
values.